
UGUSTA, Ga. (Dec. 6, 2017) – In an exciting breakthrough scientists have discovered that a common pain medication often prescribed for chronic pain can help preserve vision in a model of severe, blinding retinal degeneration. The vision preservation could activate one of the most powerful antioxidants in the human body known as Nrf2. This targets… Continue reading Augusta University Discovers How Nrf2 Activator Preserves Sight in Retinal Degeneration Model
Author: Glen Thomson - NRF2.com
New Protandim Study – International Formula Impact on Oxidative Stress

LifeVantage Corporation announced a study on Protandim which was presented at the 2014 Experimental Biology Conference held April 26-30, 2014 in San Diego, California. Experimental Biology is an annual meeting attended by more than 14,000 scientists. The theme for 2014 was “Transforming the Future through Science.” The Colorado State University study entitled Oxidative Stress is Decreased… Continue reading New Protandim Study – International Formula Impact on Oxidative Stress
Reversal of persistent fibrosis in aging by targeting nox4-Nrf2 redox imbalance – Sciencemag.org
A brand new article in Sciencemag.org presents a study demonstrating how pathological fibrosis increases with age, but how NRF2 activation in mice was able to reverse the damage and repair lung capacity and fibrosis (scar tissue) resolution. This is a promising study because the current treatments of pathological and cystic fibrosis are costly and very… Continue reading Reversal of persistent fibrosis in aging by targeting nox4-Nrf2 redox imbalance – Sciencemag.org
Inflammation: A Major Contributor to Disease and Aging. Can Nrf2 Help Reduce It?

As the boomer generation ages it is probable that there will be more posts such as the one I saw this week from a friend on Facebook. Their plea was for suggestions and recommendations for a good solution to help alleviate the pain they were experiencing from inflammation. Externally, inflammation can be recognized by redness,… Continue reading Inflammation: A Major Contributor to Disease and Aging. Can Nrf2 Help Reduce It?
Spring Cleaning Damaged Proteins with Nrf2: Good for Huntington’s Disease and Other Neurodegenerative Disorders?
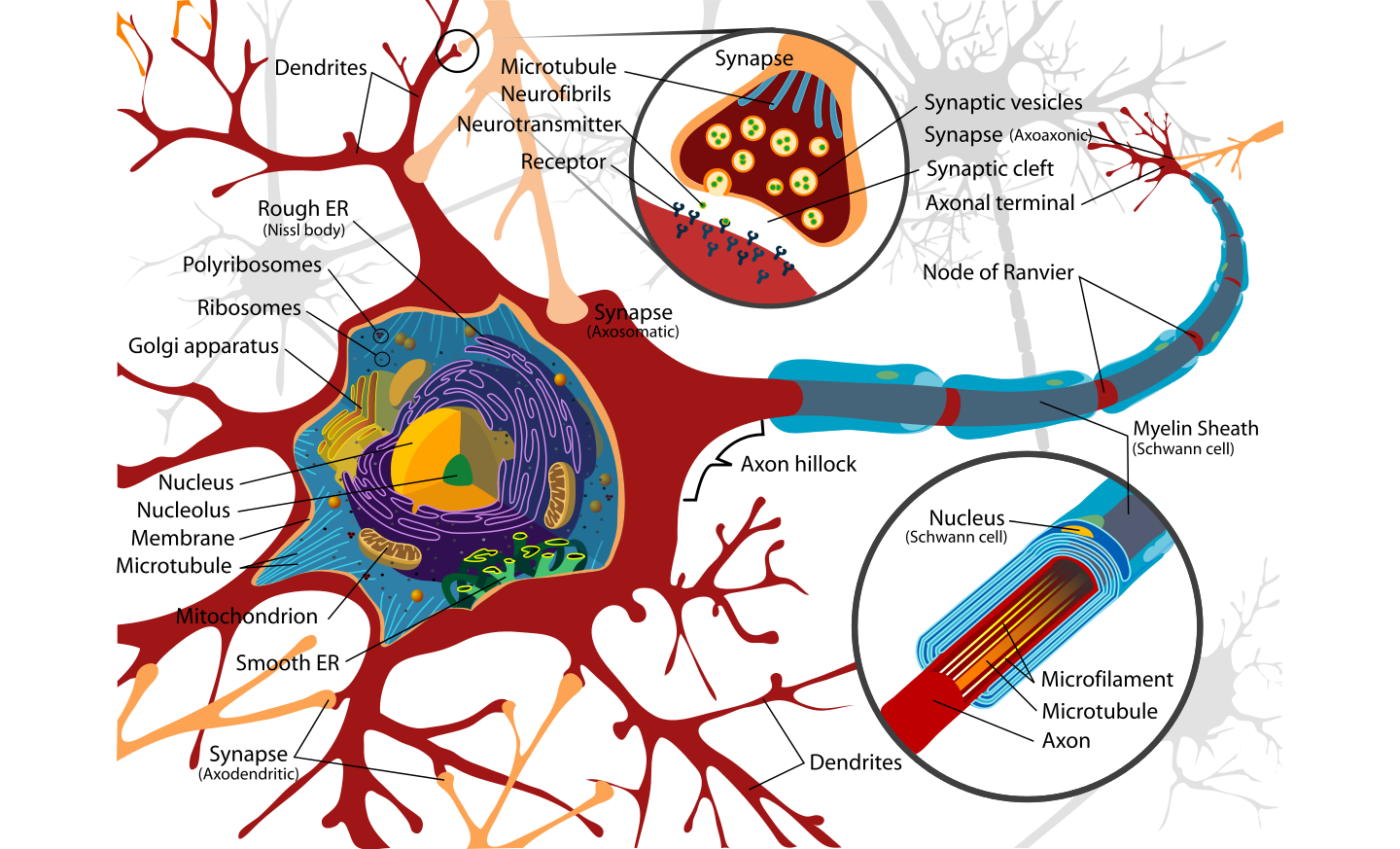
A new study supported by grants from NINDS and the National Institute on Aging as well as funding provided by the Taube/Koret Center, the National Science Foundation , the Huntington’s Disease Society of America, the Milton Wexler Award, and the Hillblom Foundation shows that activating a gene known as NRF2 helps clear damaged proteins which… Continue reading Spring Cleaning Damaged Proteins with Nrf2: Good for Huntington’s Disease and Other Neurodegenerative Disorders?
A New Nrf2 and Diabetes Study Underway
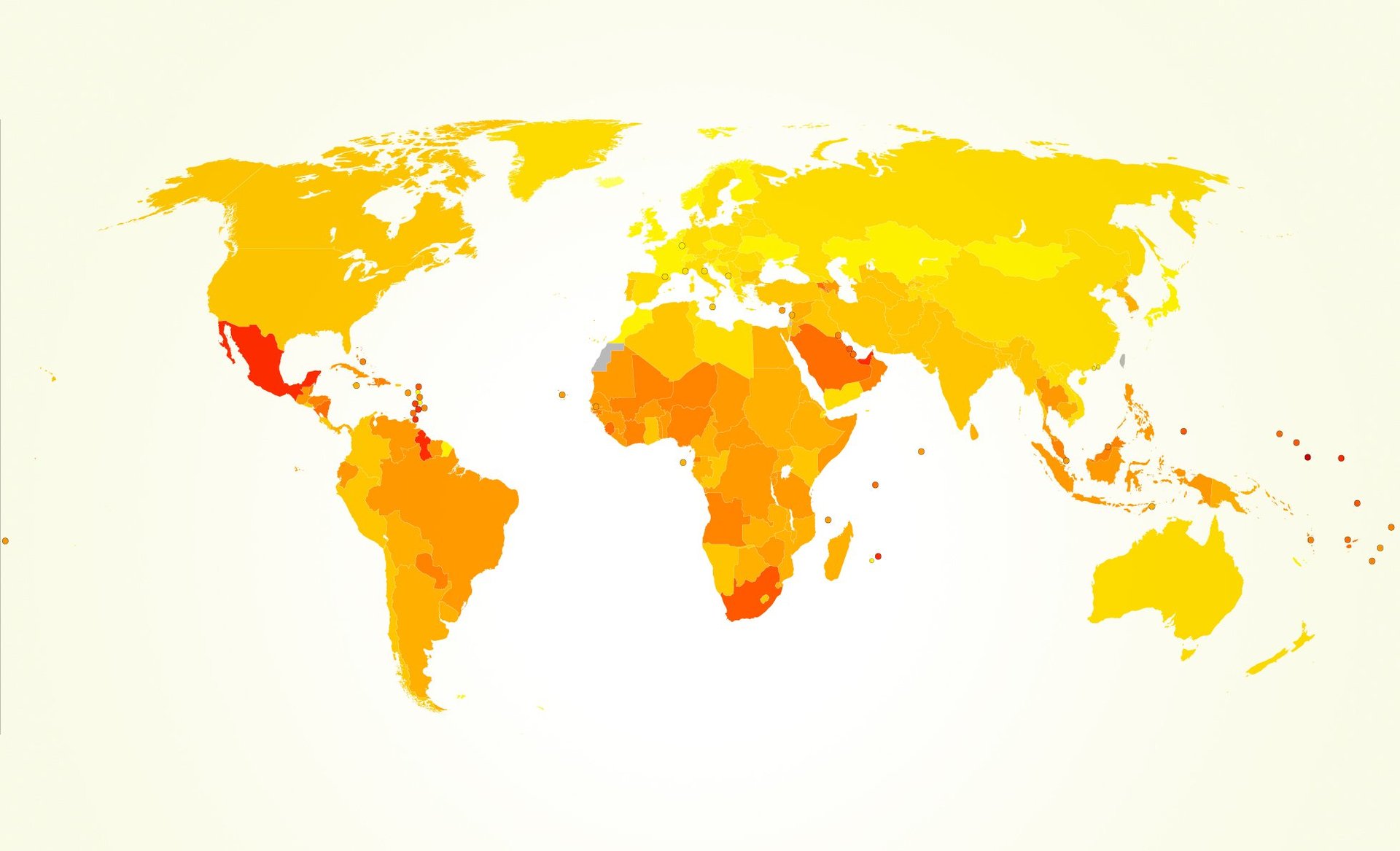
The American Diabetes Journal has details on an interesting NRF2 study currently in progress. Diabetes is a huge concern to many individuals and families. According to statistics published by the American Diabetes Association more than 25.8 million children and adults in the United States in 2010 have diabetes. That accounts for 8.3% of the population.… Continue reading A New Nrf2 and Diabetes Study Underway
New Grant Issued for Nrf2 and Epilepsy Prevention Study
Professor Matthew Walker and his fellow researchers, at University College London (UCL), have some promising early evidence in epilepsy and NRF2 research. Their research targets two approaches, 1) By targeting anti-oxidant treatments to inhibit the enzymes that produce ROS (Approach A: An attacking approach) and 2) by increasing the ability of neurons to break ROS down… Continue reading New Grant Issued for Nrf2 and Epilepsy Prevention Study
Nrf2 Benefits for Weight-loss, Obesity and Metabolic Syndrome

Obesity: A Growing Epidemic The Center for Disease Control (CDC) states that obesity is a significant public health problem. The “Healthy People” initiative shows that not one state in the United States meets the Healthy People goal of 15% body mass index (BMI). Thirty states missed the goal by a percentage by 10% or more.… Continue reading Nrf2 Benefits for Weight-loss, Obesity and Metabolic Syndrome
Angelina Jolie’s Double Mastectomy and Nrf2: Any Connections?

There has been an increased awareness and many resulting questions following Angelina Jolie’s announcement that she has had a preventative double mastectomy to prevent the further spread of cancer. The gene within her body that led to her decision is called BRCA1. BRCA1 is a tumor suppressor protein in carcinoma and normal cell types and… Continue reading Angelina Jolie’s Double Mastectomy and Nrf2: Any Connections?
Review: Nrf2, a multi-organ protector?

A classic NRF2 study entitled, “Nrf2, a multi-organ protector?” predates many of the latest studies regarding NRF2. It was however one of those landmark studies that paved the way for later studies. The study states, “Nrf2 may serve as a master regulator of the ARE-driven cellular defense system against oxidative stress.” and then goes on… Continue reading Review: Nrf2, a multi-organ protector?